J’ai loupé cette publication en août dernier. Comme dit l’auteur (anonyme), ça change tout.
« Évolution non naturelle »: preuves indiscutables de la création délibérée et systématique des variants covid en circulation
Les panels complets de « mutations de réversion » trouvés dans la circulation générale ressemblent à une expérience
26 août 2023
Le 5 août 2023, une équipe de recherche japonaise a publié une préimpression qui semble contenir les révélations les plus importantes et les plus choquantes de l’ère covid.
Atsuki Tanaka et Takayuki Miyazawa, de l’Université médicale d’Osaka et de l’Université de Kyoto, ont voulu retracer l’évolution historique du variant omicron du SARS-CoV2 en étudiant des séquences virales trouvées « dans la nature » et déposées dans des bases de données publiques.
Ils ont ainsi découvert une centaine de sous-variants omicron distincts dont l’apparition par des processus naturels n’est pas concevable. L’existence de ces variants semble apporter la preuve définitive de la création en laboratoire et de la diffusion à grande échelle de virus covid.
En outre, les variants semblent former des panels complets de mutations typiques de celles utilisées dans les expériences de « génétique inverse » pour tester systématiquement les propriétés des différentes parties des virus.
Les auteurs ont également trouvé des correspondances exactes avec des variants omicron dans des séquences provenant de Porto Rico et déposées dans des bases de données en 2020, soit plus d’un an avant l’annonce de la découverte de l’omicron en Afrique du Sud.
Associés aux observations d’un nombre invraisemblablement faible de mutations « silencieuses » dans les variants du SARS-CoV2, Tanaka et Miyazawa soutiennent que tous les variants apparus depuis l’épidémie initiale de Wuhan ne sont pas naturels et supposent qu’ils représentent un programme expérimental visant à tester les déterminants de l’infectivité et de la pathogénicité du SARS-CoV2 au sein de la population mondiale.
ADDENDUM: Le SARS-CoV2 pourrait exister en tant que « quasi-espèce virale« , c’est-à-dire
« une structure de population composée d’un très grand nombre de génomes variants, appelés spectres de mutants, essaims de mutants ou nuages de mutants ».
Cela rendrait moins surprenante l’apparition de mutations de réversion. Toutefois, cela n’expliquerait pas l’absence de mutations silencieuses dans omicron et d’autres variants (par rapport au variant original de Wuhan), ni l’absence de mutations silencieuses dans les mutants de réversion par rapport à omicron, ni la détection de séquences d’omicron en 2020 à Porto Rico.
Merci à Josh Mitteldorf de l’avoir signalé, mais il est toujours extrêmement difficile de voir comment ces observations peuvent être expliquées naturellement.
Contexte: l’évolution naturelle procède par accumulation de mutations
Avant de décrire l’étude et ses résultats, il convient de rappeler rapidement les principes de base de l’évolution des virus (et de toutes les formes de vie). N’hésitez pas à sauter cette étape si vous la connaissez déjà.
Le SARS-CoV2, comme tous les virus et organismes, se définit par son information génétique, que l’on peut considérer comme une chaîne ou une séquence de lettres. Dans la plupart des organismes, la chaîne est constituée d’ADN, mais le SARS-CoV2 et certains autres virus utilisent des chaînes d’ARN, une molécule étroitement apparentée, pour assurer cette fonction de stockage de l’information.
Le matériel génétique (ADN ou ARN) est divisé en « gènes », c’est-à-dire en séquences génétiques qui codent chacune une protéine. Les protéines sont des molécules actives qui sont synthétisées par la cellule, en utilisant le gène comme modèle. Les protéines sont également des séquences, mais leurs éléments constitutifs sont très différents. Elles n’existent pas en tant que simples chaînes de caractères qui ne font que stocker des informations, mais forment des structures 3D complexes qui ont une activité biologique.
Lorsqu’un organisme se reproduit, la séquence génétique (ADN ou ARN) est copiée et transmise à la génération suivante. Mais le mécanisme de copie est sujet aux erreurs et il arrive qu’un changement, ou « mutation », se produise. Les générations suivantes transmettent cette séquence, de sorte que les mutations s’accumulent naturellement au fil du temps.
Le diagramme ci-dessous illustre ce phénomène: une séquence d’ADN originale accumule quatre mutations successives.
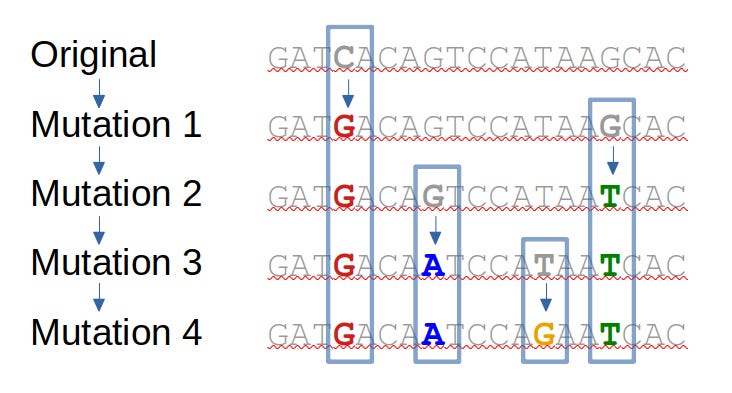
Les effets des mutations sur l’organisme déterminent leur persistance dans la nature
Il est utile de considérer qu’il existe quelques types généraux de mutation, en fonction de leurs effets sur l’organisme:
- Beaucoup n’auront aucun effet. Certaines modifications individuelles d’une lettre d’ADN/ARN ne changeront pas réellement la séquence de la protéine codée. Ces mutations « synonymes » n’ont généralement aucun effet et s’accumulent donc simplement au fil de l’évolution naturelle. Comme nous le verrons, l’absence de mutations synonymes est un signe important que l’évolution d’omicron et d’autres variants n’est pas naturelle.
- Parmi celles qui ont un effet, la grande majorité sera délétère, pour la même raison qu’une modification aléatoire d’une partie d’un système fonctionnel est susceptible de le briser. Ces mutations disparaîtront rapidement de la population.
- Il est très rare qu’une mutation provoque un changement ayant un effet bénéfique. Ces mutations proliféreront alors dans les générations futures, car les organismes qui en sont porteurs survivront et se reproduiront plus efficacement.
Ainsi, toutes les formes de vie accumulent progressivement des mutations – certaines silencieuses, d’autres bénéfiques. C’est l’évolution.
L’« évolution non naturelle » d’omicron
Examinons maintenant le cas spécifique du variant omicron du SARS-CoV2 et son évolution (présumée) à partir de la souche originale de Wuhan.
Pour cet article, nous suivrons Tanaka et Miyazawa en nous concentrant sur une seule section de la chaîne d’informations génétiques du virus – le gène codant pour la fameuse protéine de pointe. Ils étudient trois variants d’omicron officiellement reconnus – BA1, BA1.1 et BA2. Pour l’instant, nous nous contenterons d’examiner le BA1.
Comme dans toute évolution, les changements dans la protéine de pointe se produisent par l’accumulation progressive de mutations dans la séquence génétique qui la code. Dans le cas de l’omicron BA1, il y a 37 mutations non synonymes, c’est-à-dire des points où la séquence de la protéine de pointe produite est différente de celle du variant original de Wuhan.
Tanaka et Miyazawa ont voulu utiliser les bases de données publiques, dans lesquelles les chercheurs du monde entier déposent les séquences virales qu’ils ont trouvées, pour retracer l’histoire de l’évolution de la protéine de pointe de l’omicron BA1, c’est-à-dire pour répondre à la question suivante: dans quel ordre ces 37 mutations se sont-elles accumulées?
Retracer l’ordre d’accumulation des mutations omicron
Il y a deux façons évidentes de procéder: en partant de l’avant ou en revenant en arrière.
Dans l’approche prospective, on peut rechercher dans les bases de données les versions de la séquence qui présentent une seule des 37 mutations omicron, mais qui sont par ailleurs identiques à la souche d’origine. Cette seule mutation doit avoir été la première. On pourrait ensuite répéter le processus pour identifier la deuxième, la troisième, etc.
Toutefois, les variants porteurs de mutations très précoces auraient été rares dans la population mondiale du SARS-CoV2 et pourraient ne pas apparaître dans les bases de données.
Une approche plus sûre, celle adoptée par Tanaka et Miyazawa, consiste à travailler à rebours. Cela signifie qu’il faut commencer par déterminer laquelle des 37 mutations est la dernière ou la plus récente.
Pour ce faire, il faut trouver une séquence qui inclut toutes les mutations sauf une – et celle-ci doit être la mutation la plus récente.
Tanaka et Miyazawa ont donc effectué une série de 37 requêtes dans la base de données en utilisant des séquences dépourvues chacune d’une seule des mutations omicron BA1, en partant du principe que l’une des 37 séquences devrait trouver une correspondance, indiquant la dernière mutation dans la progression de l’évolution vers BA1.
Il est intéressant de s’imaginer à la place de ces chercheurs, exécutant des requêtes pour chacune des 37 mutations, se demandant peut-être laquelle d’entre elles s’avérerait être la plus récente, et obtenant la réponse… TOUTES*.
Enfin, toutes sauf une, ce qui ne fait aucune différence.
Leur cerveau a dû exploser.
Un panel de variants avec des mutations omicron BA1 inversées individuellement ne peut pas apparaître naturellement
Dans la figure ci-dessous (Fig 2A de l’article), chaque ligne représente un variant de l’omicron BA1 trouvé « dans la nature ».
Les colonnes représentent chacune des mutations omicron. Si la cellule est colorée, cela signifie que le variant porte la mutation. Les cellules blanches indiquent que la mutation est absente et que la séquence de la protéine de pointe est identique à la souche originale de Wuhan à ce stade.
Si vous pensez que le tableau a un aspect très ordonné, vous avez raison. Il montre que, pour toutes les mutations du sous-variant omicron BA1 sauf une, il existe une souche dans laquelle cette mutation – seule – est absente.
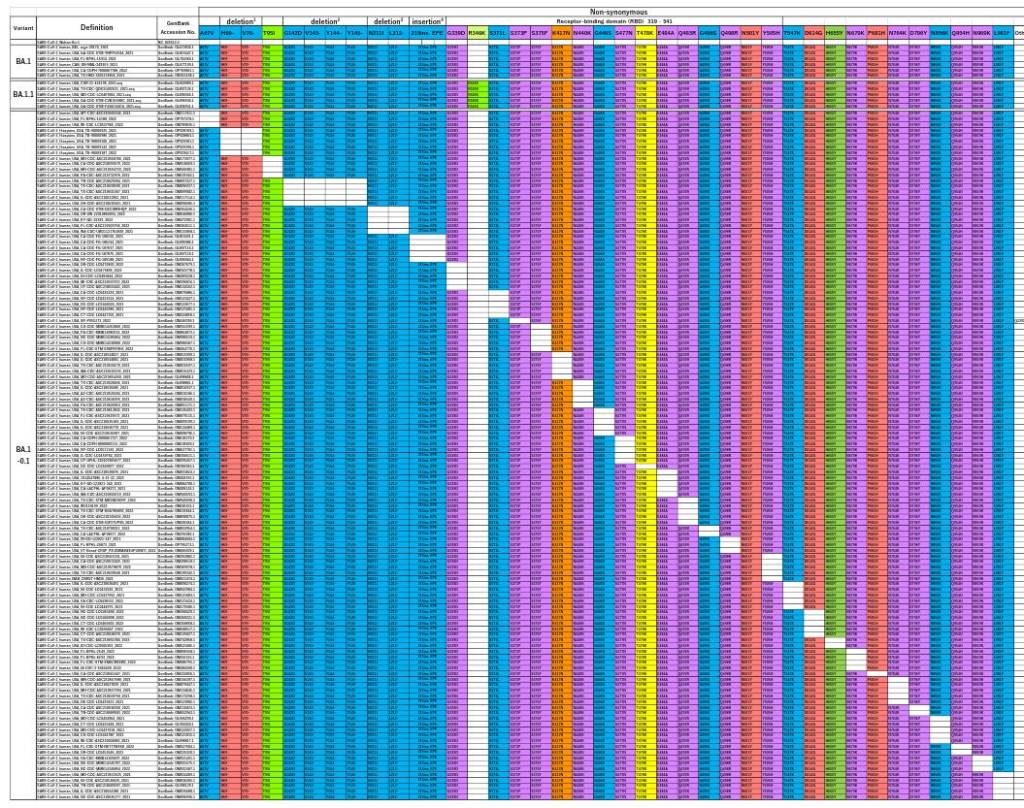
Dans l’évolution naturelle par mutation accumulée, chaque variant n’a qu’un seul parent, car il a été créé par un seul événement de mutation. Ainsi, pris au pied de la lettre, ces résultats impliquent que l’un des variants est le parent d’omicron BA1 (nous ne pouvons pas dire lequel), et que tous les autres sont des enfants.
Nous pouvons maintenant répondre à la question initiale de Tanaka et Miyazawa et décrire l’histoire naturelle de l’évolution de l’omicron BA1 qui découle de ces résultats :
- La souche BA1 officiellement reconnue se forme lorsque la dernière de ses 37 mutations se produit (nous ne savons pas laquelle);
- BA1 subit ensuite 35 changements distincts et parallèles qui inversent parfaitement l’une de ces mutations par rapport à la séquence de la souche originale de Wuhan.
C’est absurde. La réversion parfaite de mutations comme celle-ci, à une telle échelle, n’est absolument pas plausible dans le cadre d’un processus naturel.
Les variants trouvés par Tanaka et Miyazama peuvent être décrits comme un « panel » de mutations de réversion. Ce type de panel est exactement ce qu’un chercheur créerait pour tester systématiquement la contribution de différents éléments d’un virus à son activité.
On trouve également des « panels » de réversion complets pour d’autres variants officiels d’omicron
Les chercheurs ont également examiné deux autres variants omicron reconnus et largement répandus, à savoir les variants BA1.1 et BA2. Fait remarquable, ils ont trouvé les mêmes « panels » de mutations de réversion pour ces deux variants.
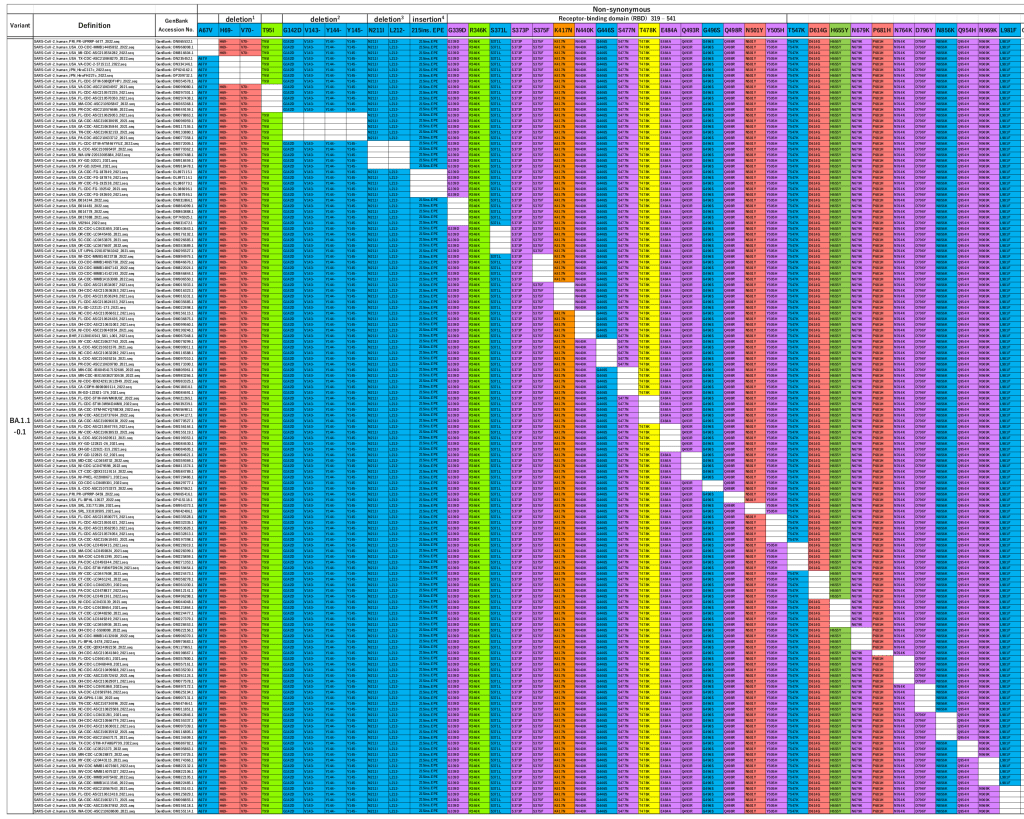
BA1.1 est très similaire à BA1 – il ne comporte qu’une seule mutation supplémentaire par rapport à la souche de Wuhan, soit un total de 38.
Lorsque Tanaka et Miyazawa ont effectué des recherches sans tenir compte de chacune de ces mutations, ils ont trouvé 37 des 38 mutations existant « à l’état sauvage ».
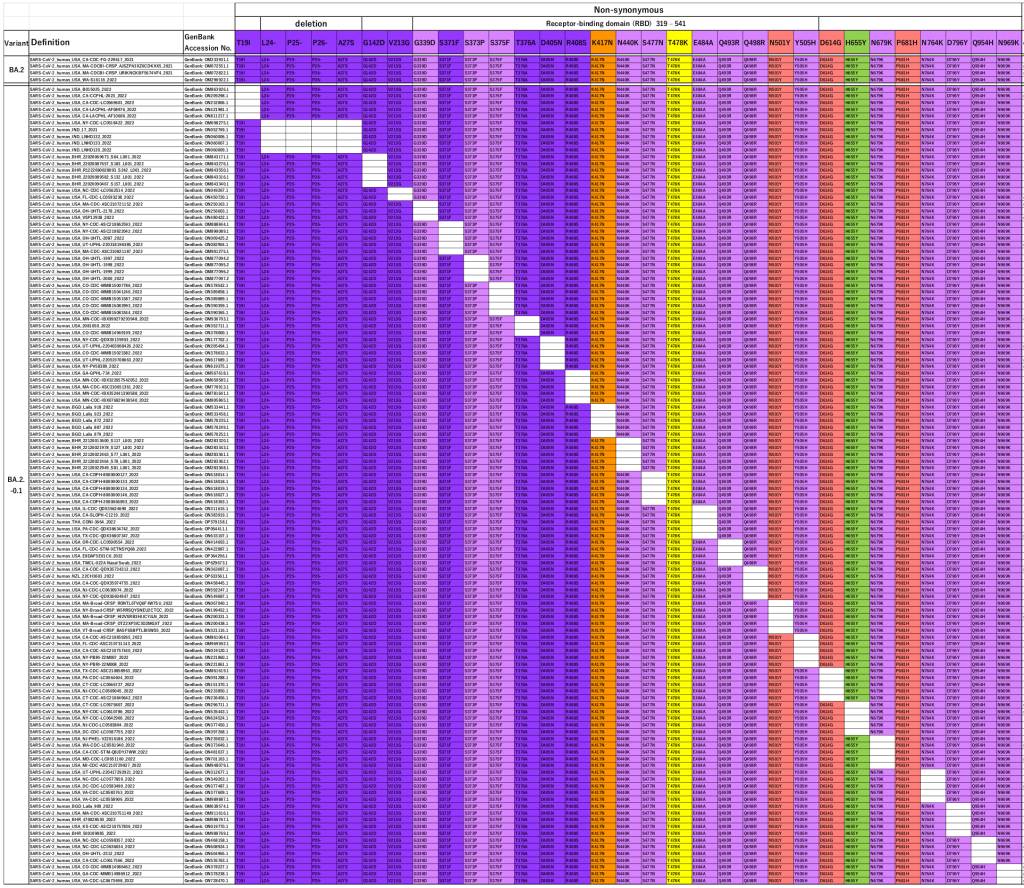
BA2 est assez différent de BA1 – ils partagent 14 mutations par rapport à la souche Wuhan, mais BA2 n’a que 17 autres mutations (différentes) pour un total de 31.
Lorsqu’ils ont recherché des variants dépourvus de ces mutations individuelles, ils en ont trouvé 29 sur les 31 « dans la nature ».
La « recombinaison », ou l’échange de matériel génétique entre différents virus, ne peut pas non plus expliquer les variants d’omicron
La discussion ci-dessus concerne l’évolution par accumulation progressive de mutations, dans laquelle chaque nouveau variant est produit par la mutation d’un seul parent.
Il existe un autre mécanisme par lequel les virus et d’autres formes de vie peuvent évoluer. La « recombinaison » implique l’échange de sections de matériel génétique entre deux variants différents. Les variants observés pourraient-ils résulter d’un échange de matériel génétique entre omicron BA1 et le virus original de Wuhan?
Tanaka et Miyazawa s’efforcent d’envisager cette possibilité, mais peuvent facilement l’exclure.
D’une part, la recombinaison nécessiterait que les virus omicron BA1 et les autres virus ancestraux soient présents dans la même cellule au même moment – car la recombinaison ne peut se produire que dans une cellule, pendant la phase de réplication du virus. Cela sera extrêmement rare compte tenu des fréquences impliquées et de la nécessité de créer autant de mutations de réversion – en particulier compte tenu de la chronologie des vagues des différents variants, comme indiqué dans l’article.
Expliquer ces réversions par la recombinaison signifierait qu’une section d’ARN dans omicron BA1 contenant la mutation à inverser devrait être proprement échangée, de sorte que les mutations situées de part et d’autre ne soient pas affectées. Il faudrait qu’il y ait deux « croisements » entre les variants, un de chaque côté de la mutation. Mais les croisements nécessitent l’alignement d’une partie de la séquence commune entre les deux souches. Pour certaines mutations, l’écart de part et d’autre de la mutation suivante n’est tout simplement pas assez grand pour permettre ces croisements, et la recombinaison est donc impossible.
La recombinaison laisserait également des traces dans les régions flanquantes du virus de part et d’autre du gène de la protéine de pointe – et celles-ci n’ont pas été trouvées.
Variants d’omicron dans des échantillons prélevés à Porto Rico – plus d’un an avant la détection officielle d’omicron
Tanaka et Miyazawa ont donc pu montrer que la recombinaison ne pouvait pas expliquer le panel de mutations de réversion qu’ils ont trouvé. Mais en envisageant cette possibilité, ils sont tombés sur d’autres éléments qui soulèvent des questions fondamentales sur l’histoire d’Omicron.
En interrogeant la base de données à la recherche de signes indiquant que la recombinaison a joué un rôle, ils ont trouvé des correspondances avec une séquence de Porto Rico soumise en 2020. D’autres recherches ont permis de trouver 29 variants attribués à Porto Rico qui correspondent exactement à omicron BA1 ou BA2, sur la base des séquences de protéines de pointe.
Toutes ces séquences ont été déposées dans la base de données en 2020, plus d’un an avant l’annonce de la détection d’omicron en Afrique du Sud en novembre 2021.
L’absence de mutations « synonymes » suggère fortement que les variants du SARS-CoV2 sont d’origine artificielle
Comme si tout cela ne suffisait pas, Tanaka et Miyazawa soulignent une autre série de preuves qui, à elles seules, suffisent probablement à conclure que les variants omicron ne sont pas naturels.
L’explication des mutations ci-dessus a souligné que, dans la nature, on s’attend à en voir
- des mutations qui ont un effet matériel et bénéfique sur l’organisme; et
- des mutations « silencieuses » ou « synonymes » qui n’affectent pas les protéines produites à partir de l’ARN/ADN et ne devraient pas modifier la capacité de reproduction de l’organisme.
Ces mutations « synonymes » qui ne modifient pas la protéine correspondante sont, comme on peut s’y attendre, beaucoup plus fréquentes que celles qui affectent la protéine elle-même. Elles n’ont généralement aucun effet sur la capacité de survie du virus et s’accumulent donc naturellement au fil du temps, parallèlement à l’acquisition de mutations non synonymes bénéfiques plus rares et plus significatives sur le plan fonctionnel.
Or, les variants officiels omicron mentionnés ici ne présentent tous qu’une seule mutation synonyme dans le gène codant pour la protéine de pointe, contre 31 à 38 mutations non synonymes.
Cela n’a aucun sens. L’évolution naturelle devrait toujours créer des mutations synonymes silencieuses à un taux plus élevé que des mutations non synonymes qui ne peuvent persister que si, contre toute attente, elles entraînent au hasard une amélioration de la conception de la protéine qu’elles codent.
Les auteurs poursuivent en soulignant que cette observation non plausible n’est pas limitée à omicron:
Il n’y avait pas de mutations synonymes dans les variants Alpha, Beta, Gamma, Delta ou Mu, [et] seulement une dans chacun des variants Lambda et Omicron.
Les panels de réversion omicron semblent faire partie d’une expérience systématique
La présence « dans la nature » de panels presque complets de réversions individuelles parfaites de pratiquement toutes les mutations dans trois lignées omicron distinctes ne peut vraisemblablement pas être naturelle. Cela ressemble plutôt à un exercice systématique de « génétique inverse » visant à tester les effets de chaque mutation omicron sur le comportement du virus.
Il est clair que tout ou partie des variants omicron ont été synthétisés dans un laboratoire d’où ils ont été libérés d’une manière ou d’une autre, dans le cadre d’un programme délibéré. Si l’on ajoute à cela l’absence de mutations synonymes dans d’autres variants, on peut en déduire que tous les variants décrits après la souche originale de Wuhan ont des origines artificielles.
Les auteurs suggèrent que les variants qu’ils ont trouvés font effectivement partie d’une expérience visant à caractériser la protéine de pointe et les effets des mutations sur le comportement du virus:
En effet, le fait que l’on n’ait pas trouvé jusqu’à présent que bon nombre des diverses mutations observées, en particulier dans les premiers variants, sont effectivement associées à une infection virale accrue (van Dorp et al, 2020) soutient l’hypothèse selon laquelle chaque variant a été synthétisé artificiellement pour identifier les acides aminés de la protéine S responsables de l’infectiosité et de la pathogénicité.
Conclusion: cela change tout
Si les observations et les déductions de cet article sont correctes – et à moins d’un pur canular, impliquant des dépôts frauduleux dans des bases de données de séquences, elles semblent l’être – alors elles fournissent la preuve indiscutable que toute l’histoire du SARS-CoV2, au moins après l’émergence de la souche d’origine, est artificielle.
Quelqu’un, quelque part, fait vraiment tout cela délibérément.
Corrections
3 septembre 2023
Certains La plupart des changements individuels d’une lettre d’ADN/ARN ne modifieront pas réellement la séquence de la protéine codée.
La majorité des mutations uniques aléatoires modifieront effectivement l’acide aminé codé. Mais l’écrasante majorité d’entre elles perturberont la protéine, de sorte qu’il est toujours vrai de dire que les mutations synonymes silencieuses devraient initialement être plus nombreuses que les mutations fonctionnelles.
